从双原子分子势能曲线获得光谱参数 (2.0版)
某些量化程序不能进行分子结构的几何优化,特别是激发态的几何优化。但是对于双原子分子来说,电子态的势能曲线形式简单,其中的大多数束缚态可以用Morse势函数表示,势函数中包含了光谱参数(Re, ωe, Te)的全部信息。因此,使用三次样条插值方法,处理用量化程序得到的双原子势能曲线,可以得到光谱参数。
计算方法
双原子分子电子态的势能曲线可以写成Morse势函数的形式:
![]() (1)
(1)
对于基态Te是0。式中
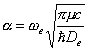 (2)
(2)
平衡位置Re可以通过求解下面方程获得:
![]() (3)
(3)
E(r)'是(1)式对r的一阶微分,Re点能量与基态平衡位置能量之差就是Te。
(1)式对r求二阶微分,其在r=Re处的值为
![]() (4)
(4)
(2)式代入(4)式,得
![]() (5)
(5)
(5)式经过变换,并经过单位换算,最后得到求ωe的公式:
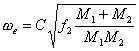 (6)
(6)
[注:计算公式为sqrt(NA*105)/2πc,其中,NA=6.022142*1023,c=2.99792458*1010 cm/s。一些文献给出的C值为1307,不准确。]
最后得到的频率ωe单位都是cm-1,M1和M2是各个原子的原子量。
因此,只要有了某个电子态的势能曲线,就可以通过以上的公式计算出光谱参数Re, ωe, Te。
应用一例
用B3LYP混合密度泛函和CEP-4G基组,计算InCl分子的基态势能曲线,并把插值拟合的结果与几何优化&频率的结果比较。密度泛函计算用Gaussian程序完成,采用默认设置。光谱常数的计算可以用商业软件Matlab或自由软件Octave完成。
第一步:获得InCl分子基态势能曲线。取一组等间隔键长r,间隔为Δr,计算各点相应的能量。把计算的键长-能量数据一一复制到MS Excel中。如表1.所示,键长取值间隔0.1 Å,因为程序用的hartree单位太大,能量已经换算到cm-1单位。
表1. B3LYP/CEP-4G计算的InCl分子基态能量(部分)
R(Å) | E(cm-1) |
2.0 | 17943.568149 |
2.1 | 9659.290704 |
2.2 | 4638.411399 |
2.3 | 1713.282007 |
2.4 | 292.456480 |
2.5 | 16.534357 |
2.6 | 436.345714 |
2.7 | 1118.609743 |
… | ... |
4.4 | 20359.116723 |
4.6 | 21776.635378 |
4.8 | 23043.651057 |
5.0 | 24154.275915 |
第二步:画图。用到的Matlab命令:
| x=[]; | 输入横轴坐标。从表1.复制第一列数据,粘贴到[]中。“;”的作用是防止回显 |
| y=[]; | 输入纵轴坐标。从表1.复制第二列数据,粘贴到[]中。 |
| plot(x,y) | 画图。 |
Octave命令:
| x=[ | 输入横轴坐标。从表1.复制第一列数据,粘贴到“[”后,然后输入“];”。“;”的作用是防止回显 |
| y=[ | 输入纵轴坐标。从表1.复制第二列数据,粘贴到“[”后,然后输入“];”。“;”的作用是防止回显 |
| plot(x,y) | 画图。 |
现在得到的势能曲线比较粗糙,无法直接用于获得光谱参数。但可以从图中初步估计平衡键长Re的位置,在2.48 Å左右。
第三步:对平衡键长Re附近的势能数据进行三次样条插值处理。插值处理区域取2.475-2.485 Å,插值间隔0.0001 Å。用到的Matlab命令:
dr=0.0001; | 产生一组等间隔的键长。在第二行命令中,第一个数值2.475是下限,第三个数值2.485是上限,dr是三次样条插值的间隔,中间用“:”隔开。后面“’”的作用是纵向排列输出结果,便于显示和复制。 |
| yi=spline(x,y,xi); | 对于势能函数y(x),在不同的键长xi处进行三次样条插值。 |
| plot(xi,yi) | 再次画图,确认能量最低点在此区域中,否则计算的光谱常数无效。 |
Octave命令:
dr=0.0001; | 产生一组等间隔的键长。在第二行命令中,第一个数值2.475是下限,第三个数值2.485是上限,dr是三次样条插值的间隔,中间用“:”隔开。后面“’”的作用是纵向排列输出结果,便于显示和复制。 |
| yi=interp1(x,y,xi,'spline'); | 对于势能函数y(x),在不同的键长xi处进行三次样条插值。 |
| plot(xi,yi) | 再次画图,确认能量最低点在此区域中,否则计算的光谱常数无效。 |
第四步:寻找势能曲线的能量最低点,求解光谱常数Re(平衡键长)和Te(基态的基准能量,或激发态的激发能)。用到的Matlab命令:
te=min(yi) | yi的最小值即为Te。 |
| re=spline(yi,xi,te) | Te对应的键长,即为Re。 |
Octave命令:
te=min(yi) | yi的最小值即为Te。 |
| re=interp1(yi,xi,te,'spline') | Te对应的键长,即为Re。 |
第五步:计算能量二阶导,即力常数f。用到的Matlab命令:
e1=spline(xi,yi,re+dr); | e1和e2是Re左右两个相邻点的能量。 |
| f=(e1+e2-te*2)/(dr*dr); | 用数值差分方法计算能量二阶导。 |
Octave命令:
e1=interp1(xi,yi,re+dr,'spline'); | e1和e2是Re左右两个相邻点的能量。 |
| f=(e1+e2-te*2)/(dr*dr); | 用数值差分方法计算能量二阶导。 |
第六步:从力常数得到振动频率ωe。用到的Matlab命令:
| m=115*35/(115+35); | 公式(6)中的InCl分子折合质量 |
| c=5.80644; | 公式(6)中的常数C。这里势能函数y(x)是cm-1-Å单位 |
| we=c*sqrt(f/m) | 得到振动频率 |
Octave命令相同。最后得到的InCl分子基态光谱参数列于表2。
表2. B3LYP密度泛函计算InCl分子基态的光谱参数。三次样条插值间隔为0.0001Å,Δr是DFT计算中分子键长取值间隔。
三次样条插值 | Gaussian结果 | 实验值 | ||
Δr=0.1 Å | Δr=0.01 Å | |||
Re(Å) | 2.4798 | 2.4793 | 2.4793 | 2.4012 |
ωe(cm-1) | 313.74 | 311.70 | 305.80 | 317.4 |
从表1可以看到,进行DFT计算的分子键长取值间隔Δr越小,结果越接近Gaussian的理论值。但是考虑到计算量的因素以及计算结果的有效数字位数,DFT计算中Δr取0.1或0.05 Å一般已经足够了。
注意:
1.这种方法不能计算反映势能曲线非谐性的ωeχe。对于浅势阱的电子态,势能曲线形状不能用Morse势函数描述。这种处理方法会使ωe和实验值有较大偏离。
2.势阱形状对Re和Te的影响不大,因此即使是浅势阱的电子态,拟合的Re和Te也是比较好的。
3.对势能曲线更高级的处理,参见LEVEL程序。
(Zork,2018.08.19更新)